 |
|
||
|
|||||||
| مشاهدة نتائج الاستطلاع: هل أنت/ أنتِ | |||
| سليم من جميع أمراض الدم الوراثية والحمدلله |
|
37 | 30.08% |
| حامل للأنيميا المنجلية |
|
10 | 8.13% |
| مصاب بالأنيميا المنجلية |
|
15 | 12.20% |
| حامل للثلاسيميا |
|
3 | 2.44% |
| مصاب بالثلاسيميا |
|
2 | 1.63% |
| حامل لأنيميا الفول |
|
10 | 8.13% |
| مصاب بأنيميا الفول |
|
7 | 5.69% |
| لا أعلم ,, فلم أخضع للفحص أبداً .. |
|
39 | 31.71% |
| المصوتون: 123. أنت لم تصوت في هذا الاستطلاع | |||
|
|
أدوات الموضوع | البحث في الموضوع | أنماط العرض |
|
#1
 01/05/2010, 06:28 PM
01/05/2010, 06:28 PM
|
||||
|
||||
   Inherited blood disorder بعد دخول البشرية الألفية الثالثة بدأت تتكشف تدريجيا اسرار عالم الوراثة المثير , واصبح هناك ضرورة لتعريف الشخص العادي عن الوراثة بصورة بسيطة وسهلة ومفهومة , فتكوين الجسم البشري بخلاياه واعضاءه يتحدد بصورة رئيسية بالتكوين الوراثي للفرد , وبالتالي فإن أي خلل في المادة الوراثية يظهر على شكل إعاقات وراثية وخلقيّة . الوسيلة الوحيدة للحد من حدوث الإعاقات الوراثية هي الوقاية منها قبل ان تحدث .. وهذا لا يأتي إلا بزيادة الوعي في المجتمع للوقاية من الأمراض والإعاقات الوراثية . زاد عدد المواضيع الإستفسارية بهذا الشأن وبشكل ملحوظ فالقسم الصحي فالسبلة وهذا ما دفعني لاختيار موضوع الحملة الثاني بتسليط الضوء على امراض الدم الوراثية بهدف زيادة الوعي في مجتمعنا العماني للوقاية من الامراض والإعاقات الوراثية وتوعية الشباب العماني بضرورة وأهمية الفحص قبل الزواج للوقاية والحد من انتشار هذه الأمراض ,, هنا سنسلط الضوء على أمراض الدم الوراثية التالية :   
6% من العمانيين يحملون جين أو مورث مرض الخلية المنجلية و 2% مرض الثلاسيميا وأكثر من 20% مرض نقص الخميرة   ملاحظه : الفحص متاح لجميع العازبين والعازبات في المؤسسات ملاحظه : الفحص متاح لجميع العازبين والعازبات في المؤسساتالصحية الحكومية والنتائج تعامل بسرية ,, لا يشترط ذهاب الطرفين , كل عازب أو عازبة مرحب بهما لعمل الفحص ... إجراءات الفحص عبارة عن ( سحب عينة دم فقط لا غير )  ,
__________________
إِدْخُـــلْ وَاسْــــتَرْخِ
اقتباس:
آخر تحرير بواسطة صرخة من صمت : 06/05/2010 الساعة 03:23 PM |
|
مادة إعلانية
|
|
#2
01/05/2010, 06:36 PM
|
||||
|
||||
|
1
 الانيميا المنجلية هي احد أنواع فقر الدم. وهي تصيب كريات الدم الحمراء.و هي من اشهر أمراض الدم الوراثية الانحلالية التي تسبب تكسر كريات الدم الحمراء-و هي اكثرها شيوعاً على مستوى العالم بشكل عام و في افريقيا و الشرق الاوسط بشكل خاص . و هناك عدة اسماء لهذا المرض ،فاضافة الى الانيميا المنجلية فانه يطلق عليها ايضا فقر الدم المنجلي او مرض المنجلية.وايا كان الاسم فالمرض واحد و ليس هناك فروق بينها. و كلمة المنجلية مأخوذه من المنجل(الذي يحصد به النبات و في بعض المناطق يطلق علية المحش)و ذلك لان كريات الدم الحمراءتحت المجهر تأخذ شكل مقوس كالمنجل او الهلال كما هو موضح فالشكل التالي ::  أسباب الخلل في الخلايا المنجلية: تفهم الخلل في كريات الدم الحمراء يجب أخذ فكرة عن تكوين هذه الخلايا وخاصة عن تكوين الهيموجلوبين خلايا الدم الحمراء تتكون في نخاع العظم ومهمتها الرئيسية نقل الأكسجين الى أعضاء الجسم المختلفة. ونقل الأكسجين يتم عن طريق ربطه على الهيموجلوبين. تركيبة الهيموجلوبين: يتكون الهيموجلوبين من الأجزاء التالية:  * الحديد * قطعة الهيم (تتكون من البروتين) * أربع قطع من البروتين المسمى بالجلوبين اثنتان من هذا الجلوبين من نوع ألفا و اثنتان من نوع بيتا وفي هذه الحالة يسمى هذا الهيموجلوبين بهيموجلوبين( أ ) قطعة الهيم وقطع الجلوبين يتم تصنيعهم في داخل الجسم عن طريق عدة مورثات (جينات) متخصصة بذلك. قطع الجلوبين من نوع ألفا يتم تصنيعها من أربع مورثات، اثنتان موجودة على كروموسوم 16 الذي ورث من الأب والاثنين الآخرين على النسخة الثانية من كروموسوم 16 الذي ورثة الإنسان من الأم. قطع الجلوبين من نوع بيتا يتم تصنيعها من مورثين اثنين واحد موجود على الكروموسوم 11 الذي ورثة الإنسان من الأب والأخرى على النسخة الثانية من كروموسوم 11 الذي ورثة الإنسان من الأم. وفي حالة الانيميا المنجلية يكون هناك عطب في كلا الجينين الذين يصنعان مادة البيتا جلوبين. أنواع الأنيميا على حسب العطب (الطفرة) في الجينات المورثة: الحامل للانيميا المنجلية: يسمى الشخص في هذه الحالة بالشخص الحامل للانيميا المنجلية وتصيب هنا الطفرة (أي العطب أو الخلل) نسخة واحدة من مورثات البيتا جلوبين فتقوم الخلية بإنتاج نوعين من البيتا جلوبين وذلك نظرا لوجود مورث أخر سليم . مورث ينتج بيتا جلوبين طبيعي ( يصنع هيموجلوبين طبيعي) و مورث يتنج بيتا جلوبين غير طبيعي. المصاب بالانيميا المنجلية: في هذه الحالة يحدث الخلل في كلا النسختان من مورثات البيتا جلوبين فتقوم الخلية بإنتاج بيتا جلوبين غيرطبيعي (والذي بتالي يصنع هيموجلوبين غير طبيعي - منجلي)
__________________
إِدْخُـــلْ وَاسْــــتَرْخِ
اقتباس:
|
|
#3
01/05/2010, 06:37 PM
|
||||
|
||||
|
العوامل والأسباب التي تؤدي الى زيادة نسبة تمنجل كريات الدم الحمراء:
وفي حالة زيادة التمنجل في الدم بسبب العوامل المذكورة أعلاه يؤدي ذلك الى إبطاء حركة الخلايا المنجلية في الدم وهذه الحالة قد تزداد سواء ً بحيث ينسد او يتجلط الدم في العروق او الشعيرات الدموية والذي يؤدي الى إقاف وصول الدم الى الاعضاء التالية مما يؤدي الى موت الخلايا و ضمورها. فاذا كان العرق يغذي العظم حدثت ألام مبرحة في تلك المنطقة و لو كان في الرئتين تحدث حالات مشابة للالتهاب الرئوي و لو كانت في المخ يمكن أن تؤدي الى جلطة في المخ والتى تسبب شللا ً نصفيا ً للمريض لا قدر الله. أعراض مرض المنجلية : بالنسبة للشخص الحامل للانيميا المنجلية لا تظهر عليه أي اعراض وقد يتم إكتشاف حمله للمرض بمجرد الصدفة وذلك عند إجراء اختبار التمنجل له والذي يجرى في العادة في كثير من البلدان بشكل روتيني قبل القيام بالعمليات الجراحية. أعراض المصاب بمرض المنجلية العامة:
أعراض لمرض المنجلية على حسب عمرالمريض: أول الأعراض الخاصة بمرض المنجلية تظهر بعد الشهر السادس من عمر المصاب: بعد الشهر السادس و خلال السنتين الاوليين من العمر وبسبب إنغلاق الشرايين في منطقة اليدين والقدمين.وذلك يؤدي الى تنفخ اليدين و القدمين والذي يكون مصحوبا بألام شديدة و بكاء متكرر. وبعض الحالات تكون مصحوبة بالتهاب فيروسي او بكتيري في الدم. بعدها تبدأ حدوث النوبات المتكررة من الالم و الناتج لانسداد العروق الدموية المغذية للعظام. وتختلف شدة هذه النوبات و عدد مرات تكرارها من شخص الى اخر. بعد سنتين الى العاشرة سنوات من العمر:
وبعد سن العاشرة من العمر تتميز بزيادة احتمال اصابة الرئتين بتجلطات نتيجة لانسددا العروق الدموية. و تظهر هذه الحالات على شكل الم في الصدر مع كحة جافة و نقص في الاكسجين وتكون الحالة مشابهة ً لإلتهاب رئوي مما يصعب عملية تشخيص الحالة. ومن المشاكل الصحية التي قد تحدث في اي عمر للشخص المصاب:
مضاعفات لمرض الأنيميا المنجلية: فقر الدم والهيموجلوبين الناتج عن تكسر وتحلل كريات الدم الحمراء المبكر يؤدي الى المضاعفات التالية:
عضلة القلب مع هبوط في القلب وعدم أنتظام في دقاته.
هرموني أن لم يعطى المريض حقنة الدسفيرال والتي تؤدي إلى التخلص من الحديد عن طريق البول.
__________________
إِدْخُـــلْ وَاسْــــتَرْخِ
اقتباس:
|
|
#4
01/05/2010, 06:38 PM
|
||||
|
||||
|
الأنيميا المنجلية الوقاية و العلاج المنجلية مرض وراثي يلازم المصاب به مدى حياته ولذلك من الأهمية القصوى أن يتبع المريض النصائح والإرشادات الطبية من طبيبه المتخصص ونذكر هنا أن العديد من مضاعفات مرض المنجلية يمكن منعها والوقاية منها وذلك عن طريق:
بعض النصائح العامة لمرضى الأنيميا المنجلية:
طرق علاج مضاعفات مرض الأنيميا المنجلية : إرتفاع الحرارة: * في هذه الحالة يجب أن يتم فحص الطفل كاملا ً وإجراء فحوصات الدم (تعداد كريات الدم, زراعة للبحث عن بكتيريا). بعض المراكز تقوم بإعطاء مضادات حيوية للأطفال الصغار بالسن عن طريق الوريد. الأطفال الكبار بالسن يتم إدخالهم للمشفى في حالة تكرار هذه الحالة. حالات الألام في العظام بسبب إنسداد العروق: * الوقاية من هذه الحالات هي العلاج الأفضل والاسهل. وفي هذه الحالات يجب حرص على تلقى الطفل الكثير من السوائل بالإضافة الى مسكنات الألام. * دواء البراسيتامول الخافظ للحرارة يخفف ايضا الالم. * ولهذا الدواء أسماء مختلفة مثل البنادول، الفيفادول، التمبرا، التيلنول. * في حالة استمرار الالم تحت علاج البراسيتمول فيجب اعطاء دواء أقوى مثل دواء الكودين فسفيت ويأخذ عن طريق وصفة من الطبيب او في المستشفى. * ومن الادوية الأقوى وفي حالة الالم الشديد فيجب اعطاء دواء المورفين او بيثيدين وذلك عن طريق الوريد. حالات الألام في الصدر: * العلاج في هذه الحالة يتشابه مع علاج ألام العظام بالإضافة الى المضادات الحيوية وإعطاء الأوكسجين. * وفي حالة عدم تحسن الحالة وإزدياد اللإتهاب الرئوي تقوم بعض المراكز بتغيير جزأ ً من الدم. في الحالات التالية يجب على أهل الطفل المريض مراجعة الطبيب أو المشفى المعالج:
هل يمكن علاج المرض نهائيا؟ المرض وراثي ومتواجد منذ ولادة المريض في نخاع العظام والعلاج المتبع هو تخفيف حدة المرض ولا يعتبر علاج شافي. لكن هناك علاج آخر كإجراء عملية استبدال نخاع العظام وهي عملية بها الكثير من المضاعفات والمخاطر وهي باهظة التكاليف ، وكذلك ليس من السهل إيجاد متبرع مناسب للمريض.
__________________
إِدْخُـــلْ وَاسْــــتَرْخِ
اقتباس:
|
|
#5
01/05/2010, 06:39 PM
|
||||
|
||||
|
2
 يعتبر مرض انيميا الفول اكثر امراض الأنزيمات انتشارا في العالم.فهو يصيب حوالي 400 مليون شخص ولو نظرنا الى التوزيع الجغرافي للمرض لوجدنا انه ينتشر في مناطق كانت موبؤه بمرض الملاريا.و مرض الملاريا من الأمراض الفتاكة و التي يسببها طفيل اسمة طفيل الملاريا.ويعيش طفيل الملاريا متطفلا على كريات الدم الحمراء فهو يستخدمها في احد اطوار حياته،و في كثير من الأحيان يؤدي الى تكسيرها وتحللها .ويبدوا ان جسم الأنسان "تأقلم" مع هذا المرض عن طريق جعل الكريات الحمراء تقاوم استيطان طفيل الملاريا فيها و ذلك بإحداث طفرة في جين انزيم G6PD فيجعل كريه الدم الحمراء تتكسر و تتحلل عند تعرضها لالتهاب بطفيل الملاريا، و بذلك يلا يستطيع الطفيل اكمال دورة حياته التي يستلزم العيش داخل كريه الدم الحمراء لبعض الوقت،و بذلك يتخلص الجسم من الملاريا بشكل فعال.وبعد ان اختفى المرض من كثير من مناطق العالم بقية الطفرة على حالها ولم يرجع الجين الى حالته السابقة.وبما ان مرض الأنيميا المنجلية و مرض الثلاسيميا ايضا يعتقد انها امراض تنتشر في المناطق الموبؤه بمرض الملاريا فإنه ليس من الغريب ان يصاب الشخص بهذه الأمراض.فبعض المصابون بمرض الأنيميا المنجلية او الثلاسيميا ايضا مصابون بمرض انيميا الفول. يعرف المرض بين الأطباء بمرض نقص خميرة (انزيم) ديهيدروجينيز الجلوكوز 6 فوسفاتي(GLOCUSE 6 PHOSPHATE DEHYDROGENASE )او بالمختصر ( G6PD). يعبتر هذا المرض مرض وراثي نتيجة لطفرة موجودة على كروموسوم اكس فلذلك يعتبر من الأمراض الوراثية التي تنتقل بالوراثة المرتبطة باالجنس.وهو في العادة يصيب الذكور و ينتقل من امهاتهم .وفي بعض الأحيان قد يظهر المرض على الاناث كما ان الذكور المصابون بالمرض ينقلون المرض ولكنهم ينقلونه الى بناتهم ولا ينقلنهوا الا ابنائهم مطلقا.ونقص اللأنزيم يجعل الكريات الدم الحمراء معرضه للتحلل والتكسر قبل موعدها المعتاد(والذي في العادة يتجاوز 100 يوم) فيؤدي الى انخفاض في الهيموجلوبين (فقر دم او انيميا)مع انتشار للمادة الصفراء تعجز عن تصفيتة الكبد بشكل سريع.هناك تفاوت كبير في السن الذي تظهر فيه اعراض المرض.فقد يظهر عند المواليد على مباشرة بعد الولادة فيكون اليرقان عندهم اعلى من المستوى المعتاد و الذي يصيب الكثير من الأطفال الطبيعين كما انه قد يحدث في أي سن و لكنه في العادة يظهر عند ما يتناول المصاب بالمرض الفول او العدس او أي نوع من البقوليات او بعد الأصابة بمرض فيروسي او عند تناول بعض من العقاقير.كما قد تظهر الاعراض من دون ان يصاب الشخص بأي مرض و من دون ان يناول أي نوع من المواد المؤكسدة كلبقوليات. ما هى علاقة المرض بالملاريا؟ مرض الملاريا من الأمراض الفتاكة و التي يسببها طفيلية المتصورة أو (البلازموديوم) .وتعيش طفيلية المتصورة متطفلة على كريات الدم الحمراء فهي تستخدمها خلال أحد اطوار حياتها للتكاثر و الاختباء، و يتأقلم جسم الأنسان مع الملاريا عن طريق جعل الخلايا الحمراء تقاوم استيطان طفيل الملاريا فيها نتيجة وجود طفرة في جين انزيم G6PD فيجعل كريات الدم الحمراء تتكسر و تتحلل عند تعرضها لالتهاب بطفيل الملاريا، و بذلك لا يستطيع الطفيل اكمال دورة حياته التي تستلزم العيش داخل الخلايا الحمراء لبعض الوقت،و بذلك يتخلص الجسم من الملاريا بشكل فعال. ماهو مرض أنيميا الفول : باثولوجيا المرض ومسبباته لنفهم مرض أنيميا الفول يجب أن نشرح وظيفة الخميرة أوالأنزيم جلوكوز- 6 - فوسفيت ديهادروجينيز (G6PD) وتأثير نقصها على أغشية خلايا الدم الحمراء دور انزيم G6PD هو انتاج مادة البنتوز من جلوكوز الفسفات السداسي و تحويل مادة النيكوتونوميد ادنين الفسفات الثنائي النووي (NADP) الى مادة النيكوتونوميد ادنين الفسفات الثنائي النووي المؤكسد (NADPH). وتكمن اهمية مادة (NADPH) في المحافظة على جعل مادة الجلوتاثيون في داخل خلية الدم الحمراء في حالة مختزلة.  وفي حالة نقص انزيم G6PD يؤدي ذلك الى نقص تكوين مادة (NADPH) وبالتالي تبقى مادة الجلوتاثيون في حالة مؤكسدة وفي هذه الحالة لا تتأثر خلايا الدم الحمراء بهذا النقص. ولكن في حالة زيادة المواد المؤكسدة مثل الفول والبقوليات و بعض العقاقير والألتهابات يؤدي ذلك الى تبلور الهيموجلوبين في داخل خلية الدم الحمراء نتيجة لعدم حمايتها من قبل مادة الجلوتاثيون وينتج عن ذلك تكسر خلايا الدم الحمراء. ولذلك فإن غالبية الأطفال المصابين بنقص الخميرة أوالأنزيم (G6PD) لاتظهر عليهم اية أعراض مرض أنيميا الفول، ولكن عند أكل الفول او بعض الأدوية، أو الإصابة ببعض الألتهابات الفيروسية يؤدي ذلك الى تكسر خلايا الدم الحمراء وظهور أعراض المرض. اين ينتشر المرض؟ ينتشر المرض في كل أنحاء العالم و لكنه يتركز في اليونان وإيطاليا ودول حوض البحر المتوسط والشرق الأوسط و جنوب آسيا ولو نظرنا الى التوزيع الجغرافي للمرض لوجدنا انه ينتشر في مناطق كانت موبؤه بمرض الملاريا. 
__________________
إِدْخُـــلْ وَاسْــــتَرْخِ
اقتباس:
|
|
#6
01/05/2010, 06:40 PM
|
||||
|
||||
|
أنيميا الفول الأســـــباب    1- تناول بعض انواع الاطعمة وعلى رأسها البقوليات بأنواعها خاصة الفول الاخضر والعدس والفاصوليا والبازلاء، ويمكن ايضاً لغبار طلع الفول ان يؤدي إلى نفس النتيجة. 2- بعض أنواع الادوية. 3- التعرض للالتهابات الفيروسية والجرثومية (مثل التهاب الكبد). 4- قد يحدث تكسر الكريات تلقائياً دون سبب واضح في بعض الحالات. أهم الادوية التي قد تؤدي إلى تكسر الكريات الحمراء عند مرضى عوزG6PD
الوراثة وأنيميا الفول  الوراثة و إنزيم G6PD يحدث مرض نقص انزيم G6PD بسبب وجود طفرة على الجين المنتج لذلك الانزيم والموجود على الذراع الطويلة كروموسوم أكس.هناك انواع كثيرة من الطفرات المؤدية لنقص او ضعف انزيم G6PD . ويعتقد انه يوجد فوق 400 نوع من الطفرات.واختلاف انواع الطفرات يفسر الى حد ما الاختلاف في الأعراض،و هناك بعض الطفرات التي تتميز بإحداث تكسر تلقائي لكريات الدم الحمراء للمواليد وهناك انواع اخرى تتميز بحدوث تكسر تلقائي مستمر للدم حتى من دون التعرض للمواد المؤكسدة كالفول..  يقع انزيم G6PD في الشريط رقم 28 من الذراع الطويلة من كروموسوم أكس X)).وبما ان المرأة لديها نسختان من كروموسوم أكس فأنها لديها أيضا نسختان من جين إنزيم G6PD نسخة أتتها من أمها و الأخرى أتتها من أبوها. وبما أن الرجل ليس لدية الا نسخة واحدة من كروموسوم أكس، فأنه لا يحمل الا نسخة وحيدة من جين الإنزيم .و بما أن الجينات قابلة للعطب فأن حدوث عطب في احد نسختي جين إنزيم G6PD في المرأة لا يؤدي في العادة للإصابة بالمرض،لان المرأة لديها نسخة إضافية على كروموسوم أكس الأخر.ولكن الرجل على خطر،فأن إصابة نسختة الوحيدة بعطب يسبب له المرض. متي يصاب الرجل بالمرض؟ يصاب الرجل بالمرض اذا كانت النسخة التي لديه من جين انزيم G6PD معطوب. و بما ان جين انزيم G6PD موجود على كروموسوم أكس و الرجل ليس لديه الا نسخة واحدة جاءته من امه فان المرض ينتقل من الام لأبنائها. و ابنائها المصابون لا ينقلون المرض لأبنائهم لانهم لا يعطون ابنائهم كرموسوم أكس فهم يعطونهم كروموسوم واي(Y). فلذلك لا ينتقل المرض من الرجل لأبنائه الذكور اطلاقاً.ولكن الرجل من المصاب من الممكن ان ينقل المرض لبناته لانه يعطيهن نسختة المعطوبة من كروموسوم أكس الوحيد عنده. متى تحمل المرأة المرض؟ تكون المرأة حاملة للمرض اذا كانت فقط احدى نسختي الجين الموجودة على كروموسوم أكس مصابة بالعطب. متى تصاب المرأة بالمرض؟ تصاب المرأة بالمرض اذا اصيبت كلتا النسختين التي لديها بالعطب.فلذلك نجد ان النسختان التي على كروموسوم أكس من الاب ومن الام معطوبة.و هذا يعني ان ابوها ايضا مصاب بالمرض و انه على اقل تقدير ان امها حاملة او مصابة بالمرض. عندما يكون الأب فقط مصاب بالمرض بينما الأم سليمة. فالمرأة اذا عطبت احدى نسختيها فانها في العادة لا تصاب المرض لان لديها نسخة اخرى. و مع ان المرأة لديها نسختين من كروموسوم أكس في كل خلية الا ان كل خلية لا يعمل فيها الا نسخة واحدة فقط و تقوم الخلية بتعطيل النسخة الثانية و ذلك لعدم حاجتها لها،و هذا شىء متوقع لان جميع خلايا الرجل تعمل بشكل طبيعي بنسخة واحدة لذلك فالله عز وجل جعل الخلية الأنثوية تعطل الكوموسوم الزائد لديها وذلك لكي لا ينتج كميه مضاعفة من البروتينات.تسمى خاصية تعطيل كروموسوم أكس في الخلية الأنثوية بتثبيط او تعطيل أكس(X inactivation ). و بما ان كل خلية فيها كوموسوم أكس من الأب و اخر من الأم فان الخلية لا تفرق بينهما فهي تقوم بتعطيل الكروموسوم بغض النظر عن مصدره؛هذه الخاصية تسمى تعطيل أكس العشوائي(Random X Inactivation). ولذلك لو نظرنا إلى جميع خلايا المرأة لوجدنا ان بعض خلاياها يعمل بها كروموسوم أكس من الأب والبعض يعمل بها كروموسوم من الأم. مشاهدة كروموسوم أكس المعطل ملتصق بجدار الخلية من الداخل ويظهر في خلايا الدم البيضاء على شكل عصيه صغيرة معروفة بعصى الطبل( Drum stick) وفي الخلايا المبطنة للفم بأجسام بار ( Bar Bodies). وبما ان التثبيط يتم بشكل عشوائي في جميع الخلايا،فاننا نجد ان حوالي 50% من الخلايا في جسم المرأة يعمل بها كوموسوم أكس مصدرة الأب و 50% يعمل بها كروموسوم أكس مصدرة الأم.. عندما تكون البنت مصابة بمرض أخر يسمي بمتلازمة تيرنر( (Turner’s syndrome: فهذا المرض يصيب البنات فقط وهو ناتج عن نقص في عدد الكروموسومات .فبدل ان يكون مجموع الكروموسومات 46 يكون لديها فقط 45 كروموسوم .والناقص هو احد نسختي كروموسوم أكس.فإذا حدث وكانت النسخة التي لديها فيها جين انزيم G6PD معطوب فأنها تصاب بالمرض وتشابه حالتها حالة الذكر المصاب بالمرض فهو لا يملك الا نسخة واحدة من كروموسوم اكس وبه نسخة معطوبة من جين انزيم G6PD.
__________________
إِدْخُـــلْ وَاسْــــتَرْخِ
اقتباس:
|
|
#7
01/05/2010, 06:41 PM
|
||||
|
||||
|
اعراض أنيميا الفول النوع الموجود في الدول العربية يسمى أنيميا الفول لحوض البحر الأبيض المتوسط (Mediterranean variant) وهو من النوع الذي يتأثر بالفول. فقر الدم و اليرقان هما أهم الاعراض وقد يحدث تكسر في كريات الدم الحمراء بشكل حاد او بشكل مزمن. ولذلك يمكن ان نقسم أعراض المرض الى هذين القسمين: التكسر الحاد لمرض أنيميا الفول يسبب تكسر الكريات الحمراء إلى انخفاض نسبة الهيموجلوبين في الدم مما يؤدى إلى:
 و تستمر الاعراض 5-6 ايام ثم تبدأ بالتحسن و أما الوفيات فإن حدثت فتكون في اليوم الاول. التكسر المزمن لمرض أنيميا الفول في بعض أنواع نقص انزيم G6PD يكون تكسر او تحلل كريات الدم الحمراء بطيء و لا تتكسر الكريات بشكل مفاجىء. هذا النوع من التكسر يسبب فقر دم مزمن وقد يكون مصحوب باصفرار بسيط في الجلد. و قد ينتج عن الإصابات المتكررة و الغير ملحوظة حصوات المرارة و تضخم الطحال. و اكثر الحالات مشاهده من أنيميا الفول عند الاطفال بين 5 اشهر حتى 5 سنوات و لم تشاهد حالات بعد سن 9 سنوات إلا نادرا و لا يزال سبب ذلك مجهولا ربما بسبب زياده تكاثر الانزيم المذكور في الكريات مع مرور الزمن . كيفية التشخيص؟
صورة دم كاملة: - فقر دم ( نقص فى الهيموجلوبين و كريات الدم الحمراء و الهيماتوكريت) - زيادة الخلايا الشبكية 15-20 %(Reticulocytes count) -جسيمات هينز(Heinz Bodies) -زيادة الكريات البيضاء إلى 40 ألف كرية لكل مم3 -نقص الهابتوجلوبين (Haptoglobin) -زيادة البيليروبين (Bilirubin) -اختبار كومب (Coomb's Test) -التحليل المؤ كد للمرض : الكشف عن نشاط الأنزيمG6PD تحليل بول: - وجود الهيموجلوبين فى حالات التكسير الشديد - وجود كميات كبيرة من اليوروبيلنوجين(Urobilinogen) مما يؤدى إلى لون غامق فى البول(برتقالى داكن).  العلاج و الوقاية المعالجة لا يوجد حتى الآن علاج شافٍ لمرض تكسر الدم الفولي و لا يمكن منعه من الانتقال من جيل لاخر، و لكن تجرى ابحاث الان على العلاج بالجينات و الخلايا الجذعية لإصلاح الجين المعطوب . *إذا كان تكسر الكريات الحمراء شديدا ًو ادى ذلك لحدوث فقر دم حاد وشديد عند المريض وهذه الحالة إسعافية تستلزم نقل الدم الاسعافي تحت إشراف طبي مع مراقبة المريض عن كثب وقد نضطر لنقل الدم اكثر من مرة، ويتم مراقبة وظائف الكلية خوفاً من حدوث الفشل الكلوي الحاد الناجم عن انحلال الدم الشديد. * راحة بالسرير أو العناية المركزة *سوائل بالوريد للتغلب على ضعف كمية البول و الدخول فى فشل كلوى * أكسجين *حبوب حمض الفوليك *حبوب الحديد *تبديل الدم (Exchange Transfusion): و هو عبارة عن إزالة معظم، أو جميع دم المستقبل، وإعطائه دم فوري بنفس الحجم الذي خسره، وهي تقنية تستعمل عند الأطفال الذين يعانون من انحلال شديد فى الدم . الوقاية ان الوقاية هي اساس تدبير هذا المرض الوراثي، فطالما كان المريض بعيداً عن الاطعمة والادوية المسببة لتكسر الدم كانت أموره سوية تماماً لذلك فإن تثقيف المريض وأهله (إن كان المريض صغيراً) من الامور الاساسية حيث لابد من التأكيد على طبيعة المرض الوراثية وانه ليس مرضاً معدياً، كما يتم تزويد المريض وأهله بقائمة الاطعمة والادوية والمواد الاخرى التي يجب ان يتجنبها لمنع حدوث تكسر الدم، ومن الامور الهامة ايضاً ضرورة تذكير الطبيب دوماً بوجود نقص انزيم G6PD عند المريض حتى يراعي ذلك عند وصف الدواء. كلمة أخيرة مرض نقص انزيم G6PD مرض شائع في منطقتنا العربية حيث أكد تقرير لمنظمة الصحة العالمية وجود مليونى طفل مصرى مصابين بأنيميا الفول، 26.4% فى البحرين،12.4% فى العراق،10% فى الأردن،27.3% فى عمان،4.5%-22% فى المملكة العربية السعودية ، وهو مرض وراثي ينقل عن طريق الوراثة المرتبطة بالصبغي الجنسي X، يؤدي المرض لحدوث انحلال دموي عند تناول بعض انواع الاطعمة (الفول، الفاصوليا) والادوية، ويظهر ذلك بالشحوب واليرقان . يجب تثقيف المريض واهله حول طبيعة هذا المرض و إنه ينبغى ممارسة الحياة بصورة طبيعية و لا يوجد اي قيود على اي انشطة او اى نوع من الرياضة و لكن لابد من الابتعاد عن كل الاطعمة والادوية والمواد الأخرى التي قد تسبب تكسر الدم لان ذلك هو السبيل الوحيد للوقاية من هذا المرض. وعلى الاطباء توخى الحذر فى وصف الادوية التى قد تسبب تكسر حاد فى كرات الدم فى مرضى انيميا الفول و ذلك عن طريق اخذ تاريخ مرضى مفصل و معرفة إذا كان اى فرد من الاسرة او الاقارب يعانى من هذا المرض.
__________________
إِدْخُـــلْ وَاسْــــتَرْخِ
اقتباس:
|
|
#8
01/05/2010, 06:42 PM
|
||||
|
||||
|
3
  الثلاسيميا من أهم أمراض الدم الوراثية الانحلالية- التي تسبب تكسر كريات الدم الحمراء- الشائعة على مستوى العالم بشكل عام وعلى مستوى منطقة البحر الأبيض المتوسط بشكل خاص ، و الثلاسيميا نوعان الألفا و البيتا وحين يأتي الحديث عن هذا المرض فإننا نقصد به (البيتا ثلاسيميا ). أصل كلمة ثلاسيميا : كلمة يونانية الأصل مشتقة من كلمة Talasso أي البحر و hemmia أي الدم تعني فقر دم منطقة البحر الأبيض المتوسط حيث أن هذا المرض عرف واشتهر في هذه المنطقة بشكل كبير ، ويعرف أيضا باسم أنيميا البحر المتوسط (Mediterranean anemia) وفي الولايات المتحدة الأمريكية كان يعرف باسم أنيميا كوليز (Cooleys anemia). وفي منطقة البحر الأبيض المتوسط كان لا بد أن يخضع كل مولود جديد بعد ولادته بشهور لهذا الفحص لمعرفة ما إذا كان حاملا أو مصابا بالبيتا ثلاسيميا . أما في منطقة الخليج العربي يحمل كل فرد من بين 20 فرد لهذا المرض أي نسبه حاملي صفة المرض في الخليج 5 % وهي نسبه لا يمكن تجاهلها حيث أنه في كل مرة تحمل فيه امرأة حاملة للمرض ومتزوجة من رجل أيضا حامل للمرض فإن احتمال إصابة الجنين تصل إلى 25% وان اكثر من 60% من أطفال هذه الأسرة السليمين أيضاً حاملين للمرض. وتكمن مشكلة المرض في عدم قدرة الجسم من تكوين كريات الدم الحمراء - التي تنقل الغذاء والأكسجين إلى مختلف أنحاء الجسم – بشكل سليم نتيجة لخلل في تكوين الهيموجلوبين( خضاب الدم) أدى إلا عدم اكتمال نضج الكريه الحمراء و تكسرها وتحللها بعد فتره قصيرة من إنتاجها (يذكر أن عمر كريه الدم الحمراء تكون 120 يوما) لذ فان المريض يحتاج إلى نقل دم بشكل دوري كل 3-4 أسابيع حسب عمره و درجه نقص الهيموجلوبين .  ينتشر المرض في عدة مناطق من العالم ولكنها تكثر في المناطق التالية: * حوض البحر الأبيض المتوسط ( اليونان، مالطا، قبرص، تركيا و إيطاليا). * منطقة الخليج العربي. * منطقة الشرق الأوسط وتشمل إيران العراق سوريا الأردن وفلسطين. *شمال أفريقيا وتشمل مصر تونس الجزائر المغرب وبعض من الدول الأفريقية الأخرى. * جنوب شرق أسيا وتشمل تايلاند، الفلبين، إندونيسيا، سنغافورة، كمبوديا، فيتنام وماليزيا. * شبه القارة الهندية. * دول أخرى كالصين، أرمينيا، جورجيا، أذربيجان.
__________________
إِدْخُـــلْ وَاسْــــتَرْخِ
اقتباس:
|
|
#9
01/05/2010, 06:42 PM
|
||||
|
||||
   المورث (الجين) المسؤول عن إنتاج مادة الألفا جلوبين: يسمى بمورث الألفا جلوبين يوجد على الكروموسوم رقم 16 ،ويوجد منه أربع نسخ (أي أربع جينات او مورثات)اثنتان منها موجودة على كروموسوم رقم 16 الأتي من الأم والاثنان الآخران موجودان على الكروموسوم الأتي من الأب. واي خلل في هذه المورثات قد يسبب مشاكل صحية حسب عدد المورثات المعطوبة او المصابة بالطفرة، واليك بعض التفصيل الألفا ثلاسيميا الساكتة Silent alpha Thalassemia): فإذا أصابت الطفرة مورث واحد فقط فأن الشخص لا يصاب بأي مشكلة صحية و تسمى هذه الحالة بالألفا ثلاسيميا الساكتة Silent alpha Thalassemia).وفي العادة لا يعرف الشخص انه حامل لمورث معطوب وقد يصعب التعرف على هذه الحالة حتى عند إجراء تحليل دم عادي(أي عدد الكريات الحمراء والهيموجلوبين )ولا يمكن الكشف علية إلا بعمل فحص لحركة الهيموجلوبين الإلكتروني في الأشهر الأولى من العمر أما بعد هذا السن فأنه يصعب اكتشاف هذا الأمر إلا بالكشف على المورثات مباشرة،وهذه غير متوفرة في الكثير من المراكز الطبية. الحامل للصفة الألفا ثلاسيميا (Alpha Thalassemia Trait) فلو أصاب العطب مورثين من هذه الأربعة فقط سمي الشخص حامل لصفة الألفا ثلاسيميا وهو لا يصاب بأية مشكل صحية ولكن قد يولد له أطفال مصابون بمشاكل صحية في الهيموجلوبين إذا كانت زوجته حاملة لنفس الصفة. مرض بالهيموجلوبين اتش : لو أصاب العطب(الطفرة)ثلاث مورثات من الأربعة يصاب الشخص بمرض يسمى بالهيموجلوبين اتش (Hemoglobin H disease) .وعند ولادة الطفل المصاب بعطب في ثلاثة مورثات من نوع ألفا يكون لدية قلة في كمية الهيموجلوبين في كريات دمهم الحمراء مع وجود هيموجلوبين غير طبيعي يسمى بهيموجلوبين بارت وهو ناتج عن اتحاد أربع قطع من جلوبين جاما .وبعد مضي بعض الأشهر(أي خلال الفترة التي تقل فيها كمية الجاما جلوبين في جسم الإنسان) يصاب الطفل بفقر دم من النوع المتوسط فيتراوح مستوى الهيموجلوبين حوالي 8-10 غرامات لكل 100 ملليتر ويكون لدية تضخم في الطحال مع يرقان . بشكل عام لا يحتاج الأطفال لنقل الدم، أما البالغين فقد يحتاجون لنقل كميات من الدم. استسقاء شديد للجنين (Hydrops Fetalis):  لو أصاب العطب(الطفرة)جميع المورثات الأربعة يصاب الجنين وهو في بطن أمه باستسقاء شديد في جميع الجسم نتيجة لوجود فقر دم حاد مع فشل في القلب كنتيجة ثانوية لنقص الهيموجلوبين في الدم. / \ / \ /   المورث (الجين) المسؤول عن إنتاج مادة البيتا جلوبين: يسمى بمورث البيتا جلوبين يوجد على الكروموسوم 11. حامل لصفة البيتا ثلاسيميا ( الثلاسيميا الصغرى) تحدث الثلاسيميا الصغرى عند وجود مورث البيتا جلوبين سليم و أخر معطوب. وفي هذه الحالة يسمى الشخص حامل للمرض(أو حامل لصفة البيتا ثلاسيميا)،ولا يعاني حامل المرض من أي مشكلة صحية إلا الهم من فقر دم طفيف ولا يحتاج إلى نقل دم و لا يستجيب للعلاج بالحديد بل يحذر من تناوله إلا إذا كان هناك نقص لمعدل الحديد في الدم.. وحسب إحصائية منظمة الصحة العالمية فان 8% من السكان حاملي الثلاسيميا الصغرى. وفي دبي 1 من كل 20 شخص وقد يجمع الفرد صفة الألفا ثلاسيميا الصغرى و البيتا ثلاسيميا الصغرى معا دون أن يؤدي ذلك إلى مرض. أما الأعراض التي قد يعاني بعض الحاملين لاحقا تكون على أنيميا شديدة للام الحامل،و بعض التغيرات في عظام الوجه و‘تضخم الطحال و أحيانا حصيات في المرارة أو تقرح في ساق القدم. حامل لصفة البيتا ثلاسيميا ( الثلاسيميا الصغرى) تحدث الثلاسيميا الصغرى عند وجود مورث البيتا جلوبين سليم و أخر معطوب. وفي هذه الحالة يسمى الشخص حامل للمرض(أو حامل لصفة البيتا ثلاسيميا)،ولا يعاني حامل المرض من أي مشكلة صحية إلا فقر دم طفيف ولا يحتاج إلى نقل دم و لا يستجيب للعلاج بالحديد بل يحذر من تناوله إلا إذا كان هناك نقص لمعدل الحديد في الدم.. وحسب إحصائية منظمة الصحة العالمية فان 8% من السكان حاملي الثلاسيميا الصغرى. وفي دبي 1 من كل 20 شخص وقد يجمع الفرد صفة الألفا ثلاسيميا الصغرى و البيتا ثلاسيميا الصغرى معا دون أن يؤدي ذلك إلى مرض. أما الأعراض التي قد يعاني بعض الحاملين لاحقا تكون على أنيميا شديدة للام الحامل،و بعض التغيرات في عظام الوجه و‘تضخم الطحال و أحيانا حصيات في المرارة أو تقرح في ساق القدم. مصاب بالثلاسيميا المتوسط تحدث الثلاسيميا المتوسطة عند وجود عطب(طفرة) في كلا مورثي البيتا جلوبين . فينتج عن ذلك نقص متوسط الشدة في كمية البيتا جلوبين المنتجة في الجسم وتؤدي في هذه الحالة إلى نقص متوسط الشدة لمستوى الهيموجلوبين في الدم فيتراوح في العادة بين 7-10 غرام لكل 100 ملليمتر ،وفي العادة لا يحتاج المريض لنقل دوري للدم ، لكنه مع التقدم في العمر و في خلال الحمل في حالة المرأة قد يحتاج إلى نقل دم وأشراف طبي بشكل منتظم. و تشكل الثلاسيميا المتوسطة من 2-10% من مرضى البيتا ثلاسيميا ،وتظهر أعراضها خلال ألسنه الثانية إلى الرابعة من عمر المريض ، و مخبرياً تظهر علامات فقر الدم خلال السنة الأولى من عمر المريض من خلال فحص الدم الروتيني.وقد يتعرض المريض لبعض المشاكل الصحية كضعف البنية وتضخم الكبد والطحال و الإصابة باليرقان(اصفرار الجلد والعينان). مصاب بالثلاسيميا (الثلاسيميا الكبرى) تحدث الثلاسيميا الكبرى أو الشديدة عند وجود عطب(طفرة) في كلا مورثي البيتا جلوبين كما هو الحال في الثلاسيميا المتوسطة ولكن نوع العطب (الطفرة)في مورث البيتا هذه المرة اشد فينتج عن ذلك نقص شديد نسبة البيتا جلوبين فينقص بذلك الهيموجلوبين إلا نسب اقل من 7 غرام لكل 100 ملليليتر نتيجة لتكسر كريات الدم الحمراء الغير طبيعية قبل انتهاء عمرها الافتراضي (120 يوم). و يحتاج المريض لنقل دوري للدم كل 3-4 أسابيع للمحافظة على نسبه عالية من الهيموجلوبين لكي ينموا الجسم بشكل صحي. ويمكن اكتشاف المرض خلال الأشهر الستة الأولى من عمر الطفل مريض بالثلاسيميا (الثلاسيميا الكبرى) + الأنيميا المنجلية في هذه الحالة يصاب المريض بمرضين في أن واحد.فيكون الشخص أما لدية مرض الأنيميا المنجلية ومرض الثلاسيميا معا أو يكون حاملا للأنيميا المنجلية مع مرض الثلاسيميا.وقد تتراوح شدة المرض وانقص الهيموجلوبين على حسب شدة الإصابة في مورث البيتا جلوبين.
__________________
إِدْخُـــلْ وَاسْــــتَرْخِ
اقتباس:
|
|
#10
01/05/2010, 06:43 PM
|
||||
|
||||
|
أعـــــــــــــــراض الثلاســيميــا تختلف أعراض المرض على حسب نوع الثلاسيميا المصاب بها الشخص. وقد لا يشعر المصاب بالمرض بأي مشكلة كما هو الحال فالشخص الحامل للألفا ثلاسيميا وتكتشف بالمصادفة عند إجراء تحليل دم لكريات الدم الحمراء والهيموجلوبين.و في المقابل قد يكون شديد كما هو الحال في البيتا ثلاسيميا الكبر في تكون الأعراض واضحة و تتفاقم المشكلة إذا لم ينقل للمريض دم بشكل دوري.كما أن المرض قد يكون شديد لدرجة أن الجنين يموت في بطن أمة أو يولد مع استسقاء في جميع الجسم نتيجة لنقص الهيموجلوبين في الدم وهبوط شديد في القلب،وهذا يحدث في الألفا ثلاسيميا التي يكون فيها جميع مورثات(جينات)الألفا هيموجلوبين معطوبة (بها طفرة مرضية).  شحوب مصحوب ببشرة داكنة مع بروز في عظام الجبهة والوجنتين والفك العلوي لطفل مصاب بالبيتا ثلاسيميا الكبرى لم ينتظم في عمليات نقل الدم  ضعف في البنية مع انتفاخ في البطن مصحوب بتضخم في الكبد والطحال وفيما يلي الأعراض التي تحدث للمصابين بمرض البيتا ثلاسيميا الكبرى إذا لم يعالج المريض:
__________________
إِدْخُـــلْ وَاسْــــتَرْخِ
اقتباس:
|
|
#11
01/05/2010, 06:44 PM
|
||||
|
||||
|
الثلاسيميا والأسبـــابـــ الثلاسيميا مرض وراثي ينتقل من الأبوين الحملين للمرض إلى بعض أطفالهم. ولكي تفهم الجزء الوراثي لهذا المرض عليك معرفة بعض الأمور المتعلقة بتصنيع الهيموجلوبين وبعض الأساسيات في علم الوراثة.   تصنيع الهيموجلوبين: يصنع الهيموجلوبين داخل خلايا الدم الحمراء والتي هي الأخرى تصّنع في نخاع العظام(مخ العظم).والهيموجلوبين عبارة عن ستة قطع مترابطة مع بعضها البعض.القطعة الأولى هي الحديد وهي تلتصق بالقطعة الثانية وهي مادة الهيم (Hem )وهاتين القطعتين محاطة بأربع قطع من البروتين المسمى بالجلوبين(Globin )اثنتان من هذا الجلوبين من نوع ألفا و اثنتان من نوع بيتا وفي هذه الحالة يسمى هذا الهيموجلوبين بهيموجلوبين ( أ ) أو (A ). هذا الترابط العجيب بين الحديد و الهيم و الجلوبين هو الذي يجعله مادة ناقلة للأكسجين في الدم. كل هذه القطع ما عدى مادة الحديد تصنع داخل الجسم البشري.وكل قطعة هي عبارة عن مادة بروتينية .وكل البروتينات في جسم الإنسان تصنع داخل الخلية.وبتحديد داخل نواة الخلية ،وبشكل أدق داخل الكروموسومات(الصبيغات) ،وبشكل اكثر دقة داخل الجين(المورث).أي أن مادة الهيم ومادة الجلوبين يصنعها عدة مورثات موجودة على الكروموسومات.ومادة الهيم يصنعها 6 مورثات موزعة على عدة كروموسومات،واي خلل في هذه المورثات يسبب مجموعة من الأمراض الوراثية ولكنها تختلف عن الثلاسيميا.مرض الثلاسيميا يحدث نتيجة خلل (طفرة) في المورثات التي تصنع مادة الجلوبين. المورثات و الجلوبين:  هناك نوعان أساسيان من مادة الجلوبين .الأولى تسمى ألفا جلوبين والأخرى تسمى البيتا جلوبين.وكما ذكرنا ففي كل هيموجلوبين قطعتان من الألفا وقطعان من البيتا جلوبين.ويصنع البيتا و الألفا جلوبين من مورثات موجود على الكروموسومات،كما هو الحال في مادة الهيم. ويوجد مورثين اثنين لتصنيع مادة البيتا جلوبين واحدة موجودة على الكروموسوم 11 الذي ورثة الإنسان من أبوه والأخرى على النسخة الثانية من كروموسوم 11 الذي ورثة الإنسان من أمة..بينما يصنع مادة الألفا جلوبين من أربع مورثات،اثنتان موجودة على كروموسوم 16 الذي ورث من الأب والاثنين الآخرين على النسخة الثانية من كروموسوم 16الذي ورثة الإنسان من أمة التي جاءت من الأم.إذا حدث خلل أو عُطب (طفرة) في أحد مورثات الألفا جلوبين يصاب الإنسان بمرض ألفا ثلاسيميا، و إذا أصاب الخلل مورث من مورثات البيتا يصاب الإنسان بمرض البيتا ثلاسيميا.هذه بشكل مبسط العلاقة بين المورثات و الثلاسيميا،ولكن الموضوع اعقد من ما ذكرنا ،ولكي تعرف المزيد تابع القراءة! البيتا ثلاسيميا (Beta Thalassemia ) : خلاصة ما ذكرنا أن هناك أربع مورثات لتصنيع مادة الألفا جلوبين، و مورثين لتصنيع مادة البيتا جلوبين.واليك الآن بعض التفصيل في هذا الأمر و لنبدأ بالبيتا ثلاسيميا.لو أصابت الطفرة(أي العطب أو الخلل) نسخة واحدة من مورثات البيتا جلوبين فأن الخلية تستمر في إنتاج البيتا جلوبين وذلك نظرا لوجود مورث أخر سليم .ولكن هناك نقص في الكمية الإجمالية المنتجة من هذه المادة ولكن هذا النقص لا يسبب مرضا ولكن يسمى الشخص الذي يحمل مورث واحدا معطوبا من مورثات البيتا بالحامل للمرض،أي انه حامل لمرض البيتا ثلاسيميا ويمكن أن يتعرف على هذه الحالة بأجراء فحص للكريات الدم الحمراء والهيموجلوبين و فحص حركة الهيموجلوبين الإلكترونية ( Hemoglobin Electrophoresis)وهي متوفرة في الكثير من المستشفيات.أما لو حدثت الطفرة في كلا النسختان فان مجوع الكمية المصنعة من البيتا جلوبين تقل بشكل كبير فيصاب الشخص بمرض البيتا ثلاسيميا وتكون شدة المرض على حسب شدة الطفرة فبعض الطفرات تسمح بإنتاج كمية معقولة من مادة البيتا جلوبين(ويسمى المرض في هذه الحالة بالبيتا ثلاسيميا الصغرى)وهناك طفرات شديدة تؤدي إلى نقص كبير وقد يكون كامل في مادة البيتا جلوبين (ويسمى المرض في هذه الحالة بالبيتا ثلاسيميا). نظرا لوجود أربع مورثات من الألفا جلوبين فأن الشخص تظهر عليه أعراض المرض على حسب عدد المورثات المعطوبة .فإذا أصيب مورث واحد فأن الشخص لا يصاب بأي مشكلة صحية(يسمى بالألفا ثلاسيميا الساكتة Silent alpha Thalassemia) وقد يصعب التعرف على هذه الحالة عند إجراء تحليل دم عادي(أي عدد الكريات الحمراء والهيموجلوبين )ولا يمكن الكشف علية إلا بعمل فحص لحركة الهيموجلوبين الإلكتروني في الأشهر الأولى من العمر أما بعد هذا السن فأنه يصعب اكتشاف هذا الأمر الا بالكشف على المورثات مباشرة. الالفا ثلاسيميا ( Alpha thalassemia): بينما يصنع مادة الألفا جلوبين من أربع مورثات،اثنتان موجودة على كروموسوم 16 الذي ورث من الأب والاثنين الآخرين على النسخة الثانية من كروموسوم 16الذي ورثة الإنسان من أمة التي جاءت من الأم.إذا حدث خلل أو عُطب (طفرة) في أحد مورثات الألفا جلوبين يصاب الإنسان بمرض ألفا ثلاسيميا. ونظرا لوجود أربع مورثات من الألفا جلوبين فأن الشخص تظهر عليه أعراض المرض على حسب عدد المورثات المعطوبة .فإذا أصابت الطفرة مورث واحد فقط فأن الشخص لا يصاب بأي مشكلة صحية و تسمى هذه الحالة بالألفا ثلاسيميا الساكتة Silent alpha Thalassemia).وفي العادة لا يعرف الشخص انه حامل لمورث معطوب وقد يصعب التعرف على هذه الحالة حتى عند إجراء تحليل دم عادي(أي عدد الكريات الحمراء والهيموجلوبين )ولا يمكن الكشف علية إلا بعمل فحص لحركة الهيموجلوبين الإلكتروني في الأشهر الأولى من العمر أما بعد هذا السن فأنه يصعب اكتشاف هذا الأمر إلا بالكشف على المورثات مباشرة،وهذه غير متوفرة في الكثير من المراكز الطبية.أما إذا أصابت الطفرة مورثين اثنين من الأربعة فان هذه الحالة تسمى بالحامل للصفة الألفا ثلاسيميا (Alpha Thalassemia Trait) وهذا الشخص أيضا لا يصب بمشاكل في الصحة وليس علية خطورة .ولكن يمكن الاشتباه في أن الشخص لدية هذا النوع من الثلاسيميا عند إجراء تحليل عادي لكريات الدم الحمراء ،ففي هذه الحالة تكون كريات الدم الحمراء اصغر من الحجم المعتاد وكمية الهيموجلوبين أيضا اقل من المعتاد.ولكن هذه العلامات التي على كريات الدم الحمراء أيضا تظهر عند بعض الأشخاص أو الأطفال المصابون بفقر الدم الناتج عن نقص تناول الحديد.لذلك يقوم الأطباء بقياس كمية الحديد في الدم فإذا كانت طبيعية فأن الاشتباه أن هذه الحالة ناتجة عن الألفا ثلاسيميا .أما إذا كان هناك نقص في الحديد فيعالج الأمر بإعطاء الحديد ثم يعاد التحليل .و إذا زال نقص الحديد واستمر صغر كريات الدم الحمراء فان هذا يعني أن الشخص كان عنده نقص في الحديد مصحوب بألفا ثلاسيميا.يكثر الاشتباه بين فقر الدم الناتج عن نقص الحديد و ألفا ثلاسيميا لذلك على العائلة تنبيه الطبيب على أن بعض أفراد العائلة لديهم ألفا ثلاسيميا لكي لا يعطى الطفل أو البالغ(كامرأة الحامل) جرعات من الحديد من دون أن يكون هناك ضرورة لهذا الأمر لأن ارتفاع كمية الحديد في الجسم مضر.أما إذا أصاب العطب(الطفرة)ثلاث مورثات من الأربعة فان الشخص يصاب بمرض يسمى بمرض الهيموجلوبين اتش (Hemoglobin H disease).وعند ولادة الطفل المصاب يكون هناك نقص في كمية الهيموجلوبين في كريات دمهم الحمراء مع وجود هيموجلوبين غير طبيعي يسمى بهيموجلوبين بارت وهو ناتج عن اتحاد أربع قطع من جلوبين جاما .وبعد مضي بعض الأشهر(أي خلال الفترة التي تقل فيها كمية الجاما جلوبين في جسم الإنسان) يصاب الطفل بفقر دم من النوع المتوسط فيتراوح مستوى الهيموجلوبين حوالي 8-10 غرامات لكل 100 ملليتر ويكون لدية تضخم في الطحال مع يرقان . بشكل عام لا يحتاج الأطفال لنقل الدم، أما البالغين فقد يحتاجون لنقل كميات من الدم.أما إذا أصاب العطب(الطفرة)جميع المورثات الأربعة يصاب الجنين باستسقاء شديد في جميع الجسم نتيجة لوجود فقر دم حاد مع فشل في القلب كنتيجة ثانوية لنقص الهيموجلوبين في الدم.ويموت الجنين إذا لم ينقل له جم في أول الحمل،و إذا ولد الطفل فانه يحتاج لنقل دم بشكل دوري نتيجة لوجود فقر دم شديد إلى متوسط الشدة.
__________________
إِدْخُـــلْ وَاسْــــتَرْخِ
اقتباس:
|
|
#12
01/05/2010, 06:45 PM
|
||||
|
||||
|
وسائل علاج البيتا ثلاسيميا
نقل دم  ينقل الدم بشكل دوري للمريض (6-8 ملليليتر دم"كريات دم حمراء" لكل كجم من وزن المريض كل 3-4 أسابيع)، ويجب ان يكون عمر الدم المنقول لا يتجاوز ال6 أيام. والنقل الدوري يحافظ على مستوى عالي من الهيموجلوبين لكي يصل الأكسجين الى اجزاء الجسم بشكل افضل فيحافظ على النمو الطبيعي للطفل، ومنع التغيرات التي تحدث في عظام الجسم،حماية القلب من مضاعفات فقر الدم، منع تضخم الكبد والطحال. ويحتاج المريض إلى كريات الدم الحمراء فقط لذا يجب ان يرشح الدم المنقول من الكريات البيضاء و الصفائح الدموية. العلاج بالديسفسرال  يترسب الحديد في جسم المريض تدريجيا مما يؤدى إلى تعطيل وظائف الخلايا وبالتالي إلى موتها وفقدان وظائفها، كما يؤدى إلى الكثير من اختلال الوظائف الهرمونية و القلبية و الكبدية و الجلدية. لذا يجب التخلص من الحديد الزائد (اكثر من 1000 مج\مل) الديسفرال عبارة عن مادة ترتبط مع الحديد في الجسم وتخرج مرتبطة بالحديد لخارج الجسم عن طريق البول، ومن هنا تأتى أهميه الديسفرال الذي يحقن عن طريق الجلد لفتره8-10 ساعات يوميا او عن طريق الوريد،والعضل إزالة الطحال للطحال دور هام في التخلص من كريات الدم الميتة نتيجة مشاركة الطحال في تعويض الجسم عن عجز نخاع العظم واستمرار تراكم الحديد بصوره كبيره يبدأ بعدها الطحال بتدمير كريات الدم الحمراء والبيضاء والصفائح الدموية ،تتفاقم حدة فقر الدم وتتناقص مناعة ومقاومة المريض ويصبح عرضة للنزف، ويتم استئصال الطحال عندما تظهر المؤشرات التالية:
زرع نخاع العظم هذه العملية تعتمد على وجود متبرع يفضل أن يكون من أشقاء او شقيقات المريض حيث يجب أجراء الفحوصات التي تؤكد من وجود التطابق النسيجي والخلوي (100%) بين المتبرع والمريض،وتزداد فرص نجاح العملية للذين لا يعانون من ترسب الحديد أو تضخم الكبد والطحال. إن رفض الانسجه عادة ما يكون اقل نسبة في الأطفال عنه في الكبار فذلك لا يحدث مباشرة بعد العملية إنما قد يحتاج إلى 3-5 سنوات يبقى فيها خطر الرفض،فالمريض بحاجة إلى متابعة دقيقه للغاية. ويتم فبل زرع نخاع العظام التخلص من نخاع المريض بتناوله الأدوية تدمر خلايا النخاع وعلى مدى 6 أيام يشعر فيها بالوهن والضعف وتحت التعقيم التام يتم شفط النخاع بواسطة حقنه خاصة وتحت التخدير العام وتستغرق هذه العملية حوالي نصف الساعة وتستكمل العملية بإعادة حقن نخاع العظام السليم إلى المريض فيما يشبه عملية نقل الدم فيجد النخاع الجديد تجويف نخاع المصاب. والمتبرع عادة لا يعاني من أي مخاطر وجسمه قادر على تعويض الكميه التي تم شفطها من نخاع العظام ولا يحتاج للبقاء في المستشفى اكثر من يوم واحد فقط.
__________________
إِدْخُـــلْ وَاسْــــتَرْخِ
اقتباس:
|
|
#13
01/05/2010, 06:51 PM
|
||||
|
||||
|
الزواج وطرق وراثة أمراض الدم الوراثية الثلاثة
هذه الصفحة توضح لنا أهمية الفحص قبل الزواج , حيث تتعرفون على طرق انتقال الأمراض الوراثية حسب الزوج والزوجة كــ (حامل+مصاب+سليم) و(حاملة +مصابة+سليمة) قبل البدئ هنا سنوضّح الفرق بين السليم وحامل المرض والمصاب به؟ 1. السليم: هو الشخص الذي لا يحمل صفة المرض ولا خطر على أطفاله من الإصابة عند زواجه بشخص مصاب أو حامل للمرض أو سليم منه. 2. الحامل للمرض: هو الشخص الذي يحمل صفة المرض ولا تظهر عليه الأعراض. وهذا الشخص يمكنه الزواج من شخص سليم وإنجاب أطفال أصحاء ولكن من الخطر زواجه من شخص مصاب أو حامل للمرض مثله حيث يكون أطفاله عرضة للإصابة بهذا المرض. 3. المصاب: هو الشخص الذي تظهر عليه أعراض المرض وهذا الشخص يمكنه الزواج من شخص سليم وإنجاب أطفال أصحاء ومن الخطر زواجه من حامل للمرض أو مصاب مثله حيث يكون أطفاله عرضة للإصابة بهذا المرض. ________ طرق توارث الأنيميا المنجلية والثلاسيميا واحدة ,,وهي كالتالي : تابع الصور :  الوالد والوالدة سليمين: في حالة زواج شخصين سليمين وكل منهم لا يحمل للثلاسيميا او الانيميا المنجلية فإن جميع أطفال هذه العائلة سليمين بإذن الله.   الوالد حامل للمرض والوالدة سليمة: في حالة زواج شخصين أحدهم حامل والأخر سليم فإن وبمشيئة الله في كل مرة تحمل فيها الزوجة تكون إحتمالية أن يكون الأطفال سالمين 50% وأن يكون الأطفال حاملون للمرض أيضا 50%.  الوالد سليم والوالدة مصابة : في حالة زواج شخصين أحدهم مصاب والأخر سليم فإن كل أطفال هذه العائلة تكون حاملة للمرض.  الوالد والوالدة حاملين للمرض: في حالة زواج شخصين كلا منهم حامل للمرض فإن أمام هذه العائلة أربع إحتمالات في كل مرة تحمل فيها الزوجة. نسبة أن يكون الطفل سليم 25%، ونسبة أن يكون الطفل مصاب أيضا 25%، ونسبة الأطفال الحاملين للمرض تكون 50%.  الوالد حامل للمرض والوالدة مصابة : في حالة زواج شخصين أحدهم حامل والأخر مصاب فإن في كل مرة تحمل فيها الزوجة تكون إحتمالية أن يكون الأطفال مصابين 50% وأن يكون الأطفال حاملون للمرض أيضا 50%.  في حالة زواج شخصين كلاهما مصاب فإن جميع أطفال هذه العائلة وللأسف الشديد يكونون أيضا مصابين بالمرض.  الزوج حامل للبيتا ثلاسيميا و الزوجة حاملة للانيميا المنجلية: لو تزوج زوجين احدهما حامل للانيميا المنجلية و الاخر حامل للبيتا ثلاسيميا فان أمام هذه العائلة أربع احتمالات في كل مره تحمل فيه الزوجة باذن الله. الاحتمال الأول من هذه الأربعة طفل سليم من المرض(أي 25% ) و الاحتمال الثاني طفل حامل للانيميا المنجلية ( أي 25% ). و الاحتمال الثالث هو طفل حامل للثلاسيميا ( أي 25% ). و الاحتمال الرابع هو طفل مصاب بالانيميا المنجلية و البيتا ثلاسيميا و يظهر المرض كانه انيميا منجلية متوسطة الشدة ( أي 25% ). طرق توارث أنيميا الفول G6PD 1- الأم سليمة و الزوج مصاب: نظراً لوجود جين انزيم G6PD على كروموسوم أكس وليس على كروموسوم واي،و نظراً الى ان الأب يعطي ابنائة الذكور كروموسوم واي ،فإن جميع ابنائة الذكور يكونوا سليمين من المرض.بينما جميع بناتة حاملات للمرض.طبعا المرأة الحاملة للمرض لا تصاب بالمرض و لا تظهر عليها اعراض المرض،و لكن فى بعض الأحيان تظهر اعراض المرض على المرأة الحاملة للمرض. 2-الأم حاملة للمرض وزوجها سليم: ان احتمال اصابة الذكور في هذه الحالة 25% في كل مرة تحمل فيها الأم. كذلك 25% من الأناث يكن حاملات للمرض.  3-الأم حاملة للمرض وزوجها مصاب: ان احتمال اصابة الذكور في هذه الحالة لا ترتفع عن الحالة السابقة فهي 25% في كل حمل.ولكن احتمال اصابة الاناث في هذه الحالة تصل الى 50%. 4-الأم مصابة بالمرض وزوجها سليم: ان احتمال اصابة الذكور في هذه الحالة هي 50% كما ان 50% من الاناث حاملات للمرض. 5- الأم و الأب مصابين بالمرض: ان احتمال اصابة الذكور والأناث في هذه الحالة 100%. 
__________________
إِدْخُـــلْ وَاسْــــتَرْخِ
اقتباس:
|
|
#14
01/05/2010, 06:52 PM
|
||||
|
||||
|
توفر الفحص قبل الزواج في جميع أنحاء السلطنة
الدكتورة نورة بنت سيف الحوسني ـ اختصاصية أولى صحة الأم والطفل عضوة الجمعية العمانية لأمراض الدم الوراثية تقول : للأسف الشديد علاج هذه الأمراض صعب جدا ويكاد يكون منعدم وكل ما يتلقاه المرضى حاليا بالمستشفيات هو علاج لتخفيف الاعراض كمسكنات الألم والمضادات الحيوية في حالة الالتهابات البكتيرية. وبما انه لا يوجد علاج لهذه الامراض فان الطريقة الوحيدة للحد من انتشار امراض الدم الوراثية هي الوقاية قبل حدوث المرض ، ولذلك وفرت وزارة الصحة بمعظم مراكز الرعايه الصحية الأولية عيادة فحوصات قبل الزواج ، وتستهدف هذه الخدمة الشباب قبل الزواج حيت تؤخذ من الشاب او الفتاه عينه بسيطه من الدم وترسل للمختبر ثم على ضوء نتيجة الفحص تقدم المشورة والنصيحة المناسبة. حول جهود السلطنة للحد من هذه الامراض قالت الدكتورة نورة : بما انه لا يوجد علاج لهذه الامراض فان الطريقة الوحيدة للحد من انتشار امراض الدم الوراثية هي الوقاية قبل حدوث المرض ، ولذلك وفرت وزارة الصحة بمعظم مراكز الرعايه الصحية الأولية عيادة فحوصات قبل الزواج ، وتستهدف هذه الخدمة الشباب قبل الزواج حيت تؤخذ من الشاب او الفتاه عينه بسيطه من الدم وترسل للمختبر ثم على ضوء نتيجة الفحص تقدم المشورة والنصيحة المناسبة. فإذا أظهرت نتيجة الفحص ان الشاب او الفتاة يحمل الجين المورث للمرض فانه ينصح ان يقوم بفحص الشخص الذي ينوي أو تنوي الزواج به كي يتاكد من خلوه من نفس الجين الحامل للمرض لان التزاوج بين شخصين يحمل كلاهما صفة المرض ينتج عنه احتمال إصابة 25% من الاطفال بالمرض بكل أعراضه. دور الجمعية العمانية وأشارت الدكتورة نورة إلى جهود الجمعية العمانية لأمراض الدم الوراثية وقالت : هي جمعية أهلية غير ربحيه هدفها تعريف المجتمع بأمراض الدم الوراثية المنتشرة ، والإرشاد إلى سبل الوقاية من هذه الأمراض المزمن ة والحد من انتشارها لدى الأجيال القادمة ، كما تهدف إلى المساعدة في رعاية المرضى وتحسين الخدمات المقدمة لهم بالتعاون مع وزارة الصحة والمؤسسات الصحية الأخرى بالسلطنة ولتحقيق تلك الأهداف تسعى الجمعية بمساعدة عدد من من المرضى واسرهم والمتخصصين من عدد من القطاعات الحكومية والأهلية إلى إقامة الندوات والمشاركة في الفعاليات الأهلية وإصدار الكتيبات والملصقات التثقيفية بهذه الأمراض والوصول إلى قطاعات واسعة من المجتمع لتعريفهم بضرورة إجراء الفحوصات المبكرة لاسيما الفحص قبل الزواج والذي يعد الطريقه الانسب للحد من هذه الأمراض.
__________________
إِدْخُـــلْ وَاسْــــتَرْخِ
اقتباس:
|
|
#15
01/05/2010, 06:53 PM
|
||||
|
||||
|
كلمة أخيرة :
أمنيتي أن أساهم ولو بالقليل في نشر التوعية بضرورة هذا الفحص الذي لا يبالي به أغلبية شبابنا ,, والدليل أيها الإحبة أننا لا نستقبل إلا عدد بسيط جدا من الراغبين بالفحص قبل الزواج , بالرغم من أن المجال مفتوح لكم في جميع أنحاء السلطنة وبشكل مجاني , ولكن لا يوجد إقبال كبير ,, فما هو السبب يا ترى ؟ الآن هو دروكم فاتخاذ القرار ,, معاـــ لنحمي أجيالنا ـــ معاً لجيل سليم ,,, تحية بحجم هذا الكون ,,, همسة : أرجــــو المشاركة فالإستطلاع ,,, وللعلم هو خاص ولا يمكن مشاهدة المصوتين . طلب: لا تنسوني من صالح الدعاء   تمّ بحمد الله المراجــــــع : كتيبات التثقيف الصحي/وزارة الصحة ويكيبيديا الموسوعة الحرة جريدة الوطن http://knol.google.com http://www.geneticblooddisorders.info/ http://www.werathah.com www.moh.om صرخة من صمت
__________________
إِدْخُـــلْ وَاسْــــتَرْخِ
اقتباس:
آخر تحرير بواسطة صرخة من صمت : 01/05/2010 الساعة 07:15 PM |
|
#16
02/05/2010, 12:08 AM
|
||||
|
||||
 
__________________
اعتذر راح اكون خارج عن السبلة لاشعار اخر لظروف خاصة غدا حين يبلى وراء الزجاج كتاب عليه اسمي الذابل مدوناتيوتنفض كفاك عنه الغبار ويخلوا بك المنزل القاحل سيلقاك وجهي خلال السطور كما يسطع الكوكب الآفل حـنـيـن خربشة عاشق تائه |
|
#17
02/05/2010, 02:13 AM
|
|||
|
|||
|
ما شاء الله عليك مشرفتنا
كفيتي ووفيتي الله يعطيك الصحه والعافيه
__________________
رفع الصور والملفات دفعه وحده 10 ملفات
www.al-a7sees.com/up نساء وبس www.al-a7sees.com/sayedati توبيكات www.al-a7sees.com/topics شات دردشة www.al-a7sees.com/chat/flashchat.php رهف الاحاسيس www.al-a7sees.com/vb |
|
#18
02/05/2010, 10:21 AM
|
|||
|
|||
|
السلام عليكم ورحمة الله وبركاته
شكرا لك اختي الفاضلة على هذا الطرح العلمي ولكن هناك مرض وراثي ربما غفلت عن ذكره وهو مرض الهيموفيليا ( النزيف الداخلي ) وهو مرض وراثي يصيب الاولاد بنسبة كبيرة اكثر من الاناث وانا بعد الشكر لله لدي ولدين مصابان بالهيموفيليا والعلاج المتبع لهم ابر تعوض المادة المفقودة في دمهم وهي العامل الثامن واسم الدواء الذي ياخذونه عن طريق ابرة في الوريد ( الفاكتر 8 ) ومن الاعراض المستمرة التي لهذا المرض نزيف داخلي يظهر بطريقة الكدمات وتنتشر باي جزء من الجسم ولا تزول الا باخذ الابرة وطبعا تاخذ وقت او نزيف خارجي اذا جرح باي شي فان النزيف لا يتوقف الا باخذ الابرة كذلك نسال الله الشفاء التام لجميع مرضى المسلمين |
|
#20
02/05/2010, 02:06 PM
|
||||
|
||||
|
اقتباس:
 الهيموفيليا [COLOR="rgb(160, 82, 45)"]هو أحد الأمراض النزفية الوراثية، ويقدر انتشاره بـ 30-40 حالة لكل مليون نسمة. المرضى المصابون بالهيموفيليا نوع " أ" يعانون من نقص جزئي أو تام أو خلل في إنتاج احد اهم بروتينات التخثر في الدم وهو العامل رقم "8" ، أما أولئك المصابون بالهيموفيليا نوع "ب" فهم يعانون من نفس المشكلة ولكن في بروتين تخثر الدم رقم "9" . وتقسم الهيموفيليا حسب شدة النقص في هذه العوامل؛ فالهيموفيليا الشديدة تشخص عندما تكون فعالية عامل التخثر -الذي حدث فيه النقص -أقل بـ 1% من المعدل الطبيعي . أما الهيموفيليا الخفيفة فتصنف عندما تكون فعالية عامل التخثر المتأثر أكثر من 5% من المعدل الطبيعي. أما الحالة المتوسطة من المرض فهي عندما تكون نسبة فعالية عامل التخثر بين 1-5% من المعدل الطبيعي . [/COLOR] 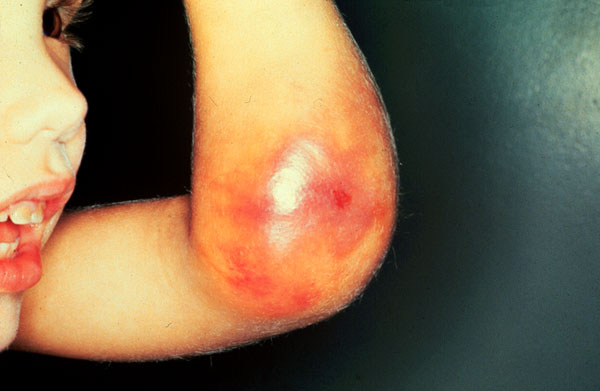 أنواع الهيموفيليا تظهر أعراض الهيموفيليا الشديدة منذ السنة الأولى في حياة الشخص المصاب إذ بمجرد ان يبدأ الطفل بالحركة تظهر علامات النزف في المفاصل وخاصة مفصل الركبة والحوض . وقد تسبب ألماً شديداً ، وفي كثير من الأحيان يحدث ضرر في المفصل مما يزيد من احتمالية حدوث إعاقة دائمة أو تشوه في المفصل والحركة. و في بعض الحالات النادرة يمكن أن يحدث النزيف كذلك في الأنسجة الداخلية أو العضلات أو القناة الهضمية الأمر الذي يعرض حياة المصاب للخطر، أما النزيف الدماغي فهو حالة طارئة جداً تهدد حياة المصاب بالخطر الشديد. وعند المصابين بالهيموفيليا، فإن أية إصابة صغيرة ، أو خلع الأسنان، أو أي إجراء جراحي بسيط أو جراحة كبرى ، هو أمر يعرض المصاب إلى احتمالية حدوث نزف لا يمكن إيقافه إن لم تتخذ الإجراءات اللازمة قبل التعرض لمثل هذه الحالات.  هيموفيليا A: [COLOR="rgb(47, 79, 79)"]وهو مرض يصيب الرجال فقط. ويمكن أن يورثه الرجل لبناته الإناث ولكن كحاملات للمرض، والإناث الحاملات للمرض يمكن أن ينقلنه وراثياً لأبنائهن الذكور، ونسبة إصابة الأبناء الذكور بهذا المرض هي 50% ، أما الإناث فنسبة كونهن حاملات ( ناقلات) للمرضى هي 50%.[/COLOR]  [COLOR="rgb(0, 100, 0)"]هيموفيليا B:[/COLOR] وهو يورث بنفس الطريقة السابقة كما في نوع A، لكن نسبة انتشاره أقل من هيموفيليا A إذ أن نسبة انتشار هيموفيليا A هي خمسة أضعاف نسبة انتشار هيموفيليا B . وبالنسبة للأعراض فهي متشابهة في النوعين ولا يمكن التمييز بينهما إلا من خلال إجراء فحص مخبري للدم لمعرفة العامل الذي حدث فيه النقص. وتسمى هيموفيليا B أحياناً بمرض كريسماس ( Christmas disease ) نسبة إلى السيد كريسماس ، أول مصاب تم تشخيصه بهذا المرض.  مرضى الهيمموفيليا والمثبطات: ويقصد بالمثبطات الأجسام المضادة المعادلة التي تتكون في جسم المصاب بالهيموفيليا بعد تلقي العلاج التعويضي بعامل التخثر المفقود أو الناقص . أما نسبة تكون هذه المثبطات (و التي تؤثر سلباً على العلاج) لدى المصابين بالهيموفيليا A هي تقريبا 10-30% و3-5% لدى مرضى هيموفيليا B. وتتكون معظم هذه المثبطات عادة في الجسم قبل سن العشرين. وتصعب معالجة الأشخاص الذين تكونت لديهم هذه المثبطات. ويمكن التنبؤ بوجود هذه المثبطات عندما يفشل برنامج العلاج التعويضي، إذ تظهر علامات عدم الاستجابة للعلاج. ويمكن معرفة مستوى تعداد هذه الأجسام المضادة في الجسم من خلال إجراء فحص خاص يقيس عددها .[/COLOR]  الهيموفيليا المكتسبة: [COLOR="rgb(160, 82, 45)"]ويحدث هذا النوع من الهيموفيليا بسبب التكون التلقائي لأجسام مضادة ( مثبطات) لعوامل التخثر رقم "9" أو "8" أما السبب في حدوث هذا النوع من المرض إما أن يكون غير معروف على وجه التحديد ، أو نتيجة للتقدم في العمر، أو الحمل، أو امرض المناعة الذاتية، أو السرطان. وعادة ما يكتشف هذا النوع من الهيموفيليا عند حدوث حالة نزف شديدة تستدعي المعالجة الطارئة، وغالباً ما يصعب معالجتها حتى مع استعمال كميات كبيرة من عوامل التخثر التعويضية.[/COLOR] [\CENTER]  علاج مرض الهيموفيليا[/CENTER] قام نوفو نورديسك بتطوير وإنتاج نوفوسيفين العامل السابع لمعالجة مرضى الهيموفيليا الذين قد تكونت في أجسامهم المثبطات ( الأجسام المضادة) . ولا تستخدم أية مواد من أصل بشري في عملية التصنيع أو في مراحل الإنتاج النهائية. ويعمل نوفوسيفن على الحد من النزف من خلال تكوين مركبات مع العامل النسجي في الموقع الذي تعرض للنزف دون التدخل أو الاعتماد على وجود عامل رقم "8" أو " 9" . إن ميكانيكية العمل هذه تفسر فعالية وتأثير نوفوسيفين في حلّ المشاكل النزفية حتى في غياب العوامل "8" و "9" منقول
__________________
اعتذر راح اكون خارج عن السبلة لاشعار اخر لظروف خاصة غدا حين يبلى وراء الزجاج كتاب عليه اسمي الذابل مدوناتيوتنفض كفاك عنه الغبار ويخلوا بك المنزل القاحل سيلقاك وجهي خلال السطور كما يسطع الكوكب الآفل حـنـيـن خربشة عاشق تائه |
|
#22
02/05/2010, 07:09 PM
|
||||
|
||||
|
اقتباس:
أتمنى تكون استفدت من الموضوع ,,,
__________________
إِدْخُـــلْ وَاسْــــتَرْخِ
اقتباس:
|
|
#23
02/05/2010, 07:13 PM
|
||||
|
||||
|
اقتباس:
__________________
إِدْخُـــلْ وَاسْــــتَرْخِ
اقتباس:
|
|
#24
02/05/2010, 07:24 PM
|
||||
|
||||
|
اقتباس:
أهلا وسهلا بك اخي الكريم .. لم أغفل المرض أستاذي ,, ولم يكن عدم ذكره تجاهلاً لوجوده في قائمة امراض الدم الوراثية ,, إنما رغبت بالتركيز على الأمراض الأكثر شيوعا فالسلطنة , وهي ما يتوفر لها الفحص قبل الزواج فالوقت الحالي ,, ولا أدري لماذا لم يتم إدراج المرض هذا للفحص ,,, قد يتم إدراجة لاحقا ,, ربما ! ,,, الله يحفظ أبنائك ويرزقك الذرية الصالحة السليمة إن شاء الله ,,, سعيدون بحضورك ,, تحية وتقدير بحجم هذا الكون ...
__________________
إِدْخُـــلْ وَاسْــــتَرْخِ
اقتباس:
|
|
#25
02/05/2010, 07:29 PM
|
||||
|
||||
|
اقتباس:
جروح قلب ,, تحية وتقدير ,, وكلمة شكر للحضور ,, في حفظ الرحمن ..
__________________
إِدْخُـــلْ وَاسْــــتَرْخِ
اقتباس:
|
|
#26
02/05/2010, 07:33 PM
|
||||
|
||||
|
تو اريد اسألك اذا اثنين يحبون بعض لدرجة الجنون وطلعت النتيجة ضدهم ينفصلو؟؟؟ صعبة ياخي 
__________________
. الــمــوت مــاهــو قــبــر ودمــوع تــســيــل الــمــوت مــوت الــقــلــب لاصــار قــاحــل كــل الــ ع ــروقــ |
|
#27
02/05/2010, 07:49 PM
|
||||
|
||||
|
اقتباس:
__________________
إِدْخُـــلْ وَاسْــــتَرْخِ
اقتباس:
|
|
#28
02/05/2010, 07:53 PM
|
||||
|
||||
|
شكراً على الموضوع الجميل والمفيد جداً بس عندي ملاحظة عن موضوع مرض الهيموفيليا ( النزيف الداخلي ) ( الناعور) ارجو مراجعة الموضوع لانه فيه اخطاء وابرزها الصورة الاول ليست لكدمة (ورم) بسبب الهيموفيليا لانه كدمة مريض الهيموفيليا تكون بلون بنفسجي الى احمر داكن وحته الصورة الثالثة ليست لها علاقة بالهيموفيليا بل صورة لعملية نقل دم والموضوع ليس به معلومات كثيرة مثل الموضوع الاول والثاني والثالث الكثيرة بالمعلومات المفيدة والدقيقة وملاحظة ثانية الهومفيليا مرض وراثي ونسبة المصابين في السلطنة كثيرة لدرجة هنك العمل قائم على انشاء جمعية خاصه لمرضى الهيموفيليا وهناك تجمع سنوي في مستشفى جامعة السلطان قابوس لمرضى الهيموفيليا ارجو من مشرفة السبلة الصحية مراجعة الموضوع للاهمية وليتعرف رواد السبلة على مرض الهومفيليا عن قرب وتفضلو بقبول فائق الاحترام والتقدير
__________________
سبحان الله والحمد لله |
|
#29
02/05/2010, 08:03 PM
|
|||
|
|||
|
أخيتي في الله صرخة من صمت
نقدم لك شكرا عاليا في جودته ساميا في مكانته غارقا في كثرته على هذه المعلومات الوافرة والوافية والمتكاملة والفحص قبل الزواج ضرورة من الضرورات يجهله الكثيرون بل قد يتجاهله البعض ويظنون ان ذلك في صالحهم في حين أن صالحهم لا يكون إلا بالفحص قبل الزواج وهناك دول بدأت لا توثق زواجا إلا إذا كان أصحابه قد اجروا فحوصات ما قبل الزواج إنها الطمأنينة التي يجب أن يكونوا عليها القادمين على الزواج حتى يطمئنوا على مستقبل أبناءهم الصحي بعيدا عن الأمراض الوراثية بارك الله فيك أخيتي ونفع بك |
|
|
 مواضيع مشابهه
مواضيع مشابهه
|
||||
| الموضوع | كاتب الموضوع | القسم | الردود | آخر مشاركة |
| : حملة سبلة عُمان للتوعية بسرطان الثدي | !بنت النور! | سبلة حواء | 0 | 21/03/2010 05:54 PM |
| أفضل حارس في العالم موسم (07-08) موضوع لنقاش | C.R.K | السبلة الرياضية | 85 | 31/07/2008 11:36 PM |
| أفضل حارس في العالم موضوع لنقاش | C.R.K | السبلة الرياضية | 1 | 04/07/2008 09:42 PM |
| الأستوديو التحليلي لمبارة ألمانيا VS أسبانيالقاء مغازلة اللقب الأوروبي | نـبض المنتدى | السبلة الرياضية | 77 | 30/06/2008 01:14 AM |
| التغطية الموحدة لأمم أوروبا 2008 أخبار × تصريحات × جدول الترتيب ×تغطية × نقاش | C.R.K | سبلة الروابط الرياضية | 108 | 21/06/2008 02:01 PM |











 النمط الخطي
النمط الخطي

